EU CTR Expert
Leer alles over de Europese Clinical Trial Regulation (EU CTR): de basisprincipes, het voorbereiden, indienen en beoordelen van onderzoeksdossiers en de regels voor de uitvoer van geneesmiddelenonderzoek in Europa in deze EU CTR Expert Training. De training bestaat uit 4 interactieve, 4 online modules en 1 toets en duurt 7,5 uur.
Doe je onderzoek in Nederland? Zie dan onze EU CTR trainingen in Nederland.
€ 95.00


Extra informatie
| Accreditatie | |
|---|---|
| Functie | Monitor/CRA, Onderzoeker, Onderzoekscoördinator, Projectmanager, Sponsor/CRO-medewerkers |
| Taal | |
| Niveau | |
| Fase | Afronding en archivering, Basis, Beoordelingsproces, Indiening, Opzet, Start onderzoek, Uitvoer, Voorbereiding |
| Regio | |
| Wetten en regels |
MEER DAN 5 LICENTIES NODIG?
LEERDOELEN
- Hoofdlijnen van de EU CTR (EU Verordening voor Klinisch onderzoek met Geneesmiddelen voor menselijk gebruik)
- Wanneer wordt de EU CTR van kracht en hoe ziet de overgangsregeling eruit
- Tijdlijnen van het indienings- en beoordelingsproces binnen de EU CTR
- Begrijp de veranderingen en wees voorbereid op de invoering van de EU CTR eind 2021
- Inzicht in de rollen en tijdlijnen die betrokken zijn bij het medisch-ethische beoordelingsproces van indieningsdossiers
- Hoe indieningsdossiers te ontwikkelen in overeenstemming met de nieuwe EU CTR-verordening
- Vereisten aan de opzet van indieningsdossiers volgens de EU CTR
- De nieuwe tijdlijnen voor toetsingscommissies en indieners
- Communicatie met de toetsingcommissie en tijdslijnen voor beantwoorden van vragen/aanleveren aanvullende informatie
- Hoe voeren we klinisch geneesmiddelenonderzoek in Europa uit volgens de EU CTR?
- Publicatie en openbaarmaking van de samengevatte onderzoeksresultaten
- Proces van indienen en beoordelen van amendementen en definitie substantiële amendementen volgens de EU CTR
- Eisen aan meldingen van AEs, SAEs, SUSARs en ernstige inbreuken.
BESCHRIJVING
EU CTR staat voor European Clinical Trial Regulation. De verordening is de Europese wet die de opzet, indiening, uitvoer en afronding van klinisch geneesmiddelenonderzoek in de Europese Unie (EU) regelt.
De EU CTR is voor iedereen die betrokken is bij klinisch geneesmiddelenonderzoek in Europa en draagt bij aan een betere samenwerking tussen alle betrokken partijen, zoals indieners, beoordelaars van toetsingscommissies en de bevoegde instanties van de verschillende Europese lidstaten.
De EU CTR expert training geeft je allesomvattend inzicht in het werken volgens de eisen van deze regelgeving in Europese geneesmiddelenonderzoek, waarmee je in staat bent deze overwegingen mee te nemen in strategische beslissingen. Van onderzoeksontwerp, tot indieningsstrategieën en van goedkeuring tot uitvoering en publicatie van onderzoeksresultaten.
Het is een ideale e-learning voor professionals die verantwoordelijk zijn voor productontwikkeling en -management, onderzoeks- en wetenschapscoördinatoren, hoofdonderzoekers (bedrijfs- en onderzoeker geïnitieerd onderzoek), medisch directeuren, managers van ClinOps afdelingen, Project Managers, CRAs, Monitors en de Regulatory Affairs afdeling, verantwoordelijk voor het indienen van dossiers.
De online training duurt 7,5 uur en bestaat uit de modules Basis EU CTR, Voorbereiding, Indiening en Beoordeling, Tijdens en na de studie en een kennistoets. Na voltooiing ontvang je een online EU CTR certificaat.
TRAININGSINHOUD
Module: Basis
- Doelstelling EU CTR en verschil tussen Directive (richtlijn) en Regulation (wet).
- Op welke typen onderzoeken is de EU CTR van toepassing
- Globale hoofdlijnen van de EU CTR
- Ambities van Nederland mbt de EU CTR
- Kansen, voordelen en moeilijkheden van de EU CTR en het indienen in de EU-portal
- Wanneer wordt de EU CTR van kracht en hoe ziet de overgangsregeling eruit
- Tijdlijnen van het indienings- en beoordelingsproces binnen de EU CTR
- Verschil in processtappen tussen indiening van een nationaal onderzoek en een internationaal onderzoek
- Resultaten van de valideringsfase, beoordeling deel 1 en beoordeling deel 2
- Verschil tussen het indieningsproces van investigator initiated onderzoek en sponsor (farma/biotech) initiated onderzoek
Module: Voorbereiding voor indiening
- Reikwijdte van de EU CTR en internationale verschillen
- Voorbereiden van indiening volgens de EU CTR en van indiening volgens de EU CTR
- Rollen en verantwoordelijkheden bij voorbereiding van indiening, incl legal representative
- Voor- en nadelen verschillende indieningsstrategieën
- Overgangsperiode
- Kent de functies en werking van de EU-portal (CTIS)
- Inhoud indieningsdossier deel 1 en van deel 2, incl protocol, CTA, pediatrisch onderzoeksplan
- Indieningsproces investigator geïnitieerd onderzoek en sponsor (farma/biotech) geïnitieerd onderzoek
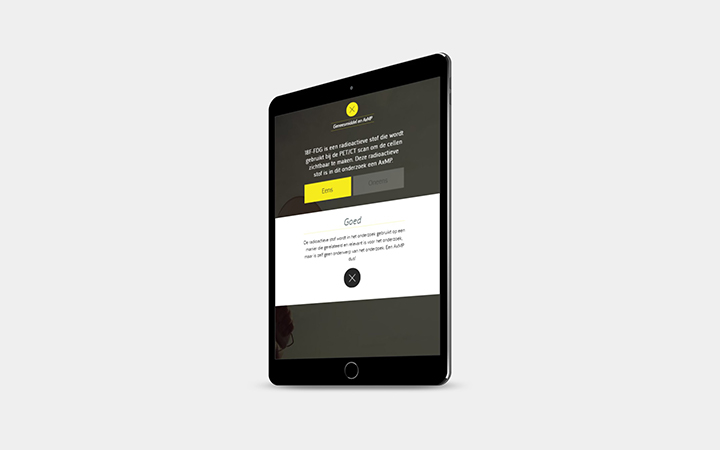
- Definitie en criteria voor gebruik en documentatie van auxiliary medicinal product (AxMP)
- Eisen aan indieningsdossier bij onderzoek met wilsonbekwame personen, klinische proeven in noodsituaties, of onderzoek met zwangere of borstvoeding gevende vrouwen
- Eisen aan productie en labelen voor geneesmiddelen voor onderzoek (IMP’s)
- Eisen aan (aansprakelijkheids-) verzekering
Module: Indiening en beoordeling
- Stappen, communicatie, resultaat en tijdslijn van het validatieproces van deel 1 (multinationaal) en deel 2 (multinationaal)
- Indienen in de EU portal (CTIS)
- Bepaling van welke lidstaat RMS en bereidheid
- Aandachtspunten beantwoorden van de vragen en het aanvullen van het onderzoeksdossier
- Samenstelling beoordelingscommissie
- Beoordelen of een onderzoek binnen de reikwijdte van de EU CTR valt
- Beoordelen van de volledigheid en geschiktheid van het onderzoeksdossier
- Geconsolideerde en gecoördineerde beoordeling
- Hoe komt een besluit tot stand
- Optimaliseren van de opstart van een studie
- Eisen aan de start van een goedgekeurde studie
Module: Tijdens en na de studie
- Stappen en bijbehorende communicatie/resultaat van het indienen en beoordelen van een aanvraag voor een substantiële wijziging die onder deel 1/ deel 2 valt.
- Definitie en voorbeelden substantiële wijziging en indiening substantiële wijziging
- Onder welke voorwaarden kan een verzoek tot substantiële wijziging ingediend worden en wanneer is er sprake van een gemixte indiening
- Eisen zijn aan de meldingen van AEs en SAEs aan de sponsor; in welke gevallen van de standaard mag worden afgeweken en hoe dit moet worden gedocumenteerd
- Wanneer een DSMB moet worden ingesteld indien bepaalde SAEs niet aan de sponsor worden gemeld
- Eisen zijn aan de meldingen van SUSARs
- Definitie, voorbeelden en procedure bij serious breach
- Stappen en bijbehorende communicatie/resultaat van einde van een klinische proef, tijdelijke stopzetting, voortijdige beëindiging en indiening van resultaten
- Belang van transparantie en documentatie rond archiveren
- Inhoud, publicatie en openbaarmaking van wetenschappelijke samenvatting en lekensamenvatting
- Eudravigilance databank voor ongeblindeerde SUSAR meldingen en jaarlijkse rapportage
- Bewaartermijnen van documentatie
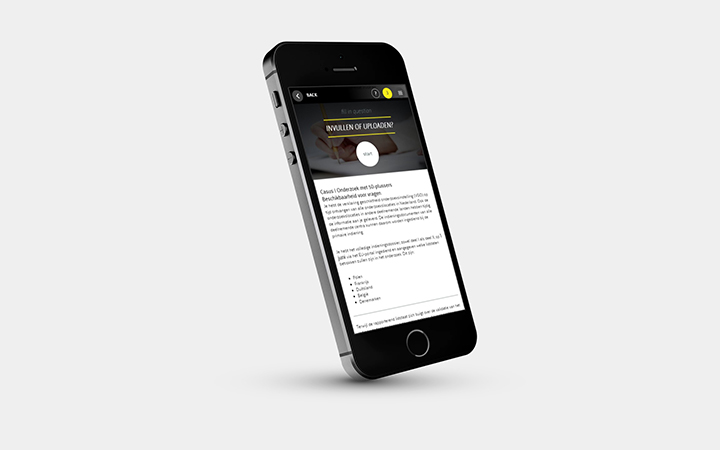
Module: Kennistoets
- 40 vragen
- Beschikbaar in Nederlands en Engels
- Feedback per vraag (achteraf)
- 3 pogingen
- Online certificaat bij een score van 80 of hoger
Gerelateerde producten
-

Introductie GCP Training voor de Afronding van Klinisch Onderzoek in Europa
€ 65.00 -

Basisprincipes ICH GCP Training voor de Start en Uitvoer van Internationaal Klinisch Onderzoek
€ 99.00 -

Introductie GCP Training voor de Indiening van Klinisch Onderzoek in Europa
€ 65.00 -

Introductie WMO GCP Training voor de Uitvoer van Klinisch Onderzoek in Nederland
€ 65.00
ALTIJD EN OVERAL LEREN
LEER VOORTDUREND
VERDIEN JE GCP-CERTIFICAAT
Toon beoordelingen in alle talen (1)
Je moet ingelogd zijn om een beoordeling te plaatsen.
KEN JE IEMAND DIE DEZE TRAINING LEUK ZOU VINDEN? DEEL HET MET ZE…
KEN JE IEMAND DIE DEZE TRAINING LEUK ZOU VINDEN? DEEL HET MET ZE:
-

Expert ICH GCP Training voor Sponsors in Internationaal Klinisch Onderzoek
€ 99.00 -

GCP Refresher (AMG) Voor Klinische Geneesmiddelenonderzoeken In Duitsland
€ 199.00 -

Expert ICH GCP Training voor Sites in Internationaal Klinisch Onderzoek
€ 99.00 -

MDR Training voor Klinisch Onderzoek in Europa
€ 199.00


beoordelingen